Department of Physical Organic Chemistry
Non-covalent Interactions Laboratory
Short URL for media go.spbu.ru/rgtolstoyp

Group members
 |
Dr. Peter M. TolstoyGroup HeadProfessor http://www.researcherid.com/rid/J-2966-2013 This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Dr. Alexander S. AntonovAssociate Professor https://orcid.org/0000-0001-7047-789X This email address is being protected from spambots. You need JavaScript enabled to view it. room 2123 |
 |
Dr. Elena Yu. Tupikinahttps://orcid.org/0000-0002-0998-8348 This email address is being protected from spambots. You need JavaScript enabled to view it., This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Dr. Vladimir Yu. MikshievThis email address is being protected from spambots. You need JavaScript enabled to view it., This email address is being protected from spambots. You need JavaScript enabled to view it. room 2135 |
 |
Dr. Valeriia V. Mulloyarovahttp://orcid.org/0000-0002-5688-2431 This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Victor G. BardakovPhD student http://orcid.org/0000-0003-2752-1976 This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Mikhail A. KostinPhD student (faculty of physics) This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Alexandra M. PuzykPhD student This email address is being protected from spambots. You need JavaScript enabled to view it. |
Students
 |
Omar AlkhuderMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Valerii V. VerkhovMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Mark V. KaplanskiyBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Valerii V. KarpovMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Vladislav O. KorostelevMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Danil V. KrutinBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Stepan A. MeshalkinBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Denis V. SolovevBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Ilja A. TatarinovBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Anna A. TitovaBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Dmitrii O. TolochenkoMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Edem R. ChakalovMaster student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Daniil A. ShitovBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Artyom A. Yakubenkohttp://orcid.org/0000-0001-5193-3298 This email address is being protected from spambots. You need JavaScript enabled to view it. |
Alumni
 |
Ivan S. GibaPhD in physics
|
 |
Alexandra A. EfimovaMaster of physics |
 |
Mikhail A. Zolenko |
 |
Andrey A. SudarkinBachelor student This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Daria O. UstimchukBachelor of Chemistry This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Ivan S. Chunarev |
 |
Alexei S. Ostras'Bachelor of Chemistry This email address is being protected from spambots. You need JavaScript enabled to view it. |
 |
Anastasia V. NamMaster of Chemistry This email address is being protected from spambots. You need JavaScript enabled to view it. |
Collaborations
| Colleague | Joint Publications |
|---|---|
|
Dr. Ilja G. Shenderovich Department of Chemistry, University of Regensburg, Regensburg, Germany |
K. Jóźwiak, A. Jezierska, J.J. Panek, E.A. Goremychkin, P.M. Tolstoy, I.G. Shenderovich, A. Filarowski, “Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates”, Molecules 2020, 25, 4720. DOI: 10.3390/molecules25204720. E.Yu. Tupikina, M. Sigalov, I.G. Shenderovich, V.V. Mulloyarova, G.S. Denisov, P.M. Tolstoy, “Correlations of NHN hydrogen bond energy with geometry and 1H NMR chemical shift difference of NH protons for aniline complexes”, J. Chem. Phys. 2019, 150, 114305. DOI: 10.1063/1.5090180. |
|
Prof. Dr. Aleksander Filarowski Department of Chemistry, Wroclaw University, Wroclaw, Poland |
Ł. Hetmańczyk, P. Szklarz, A. Kwocz, M. Wierzejewska, M. Pagacz-Kostrzewa, M.Ya. Melnikov, P.M. Tolstoy, A. Filarowski, “Polymorphism and Conformational Equilibrium for Nitro-Acetophenone in the Solid State and Under the Matrix Condition”, Molecules 2021, 26, 3109. DOI: 10.3390/molecules26113109. Ł. Hetmańczyk, E.A. Goremychkin, J. Waliszewski, M.V. Vener, P. Lipkowski, P.M. Tolstoy, A. Filarowski, “Spectroscopic identification of hydrogen bond vibrations and quasi-isostructural polymorphism in N-salicylideneaniline”, Molecules 2021, 26, 5043. DOI: 10.3390/molecules26165043. K. Jóźwiak, A. Jezierska, J.J. Panek, E.A. Goremychkin, P.M. Tolstoy, I.G. Shenderovich, A. Filarowski, “Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates”, Molecules 2020, 25, 4720. DOI: 10.3390/molecules25204720. |
|
Prof. Dr. Alexander Pozharskii Department of Organic Chemistry, Southern Federal University |
P. Piękoś, A. Jezierska, J. Panek, E.A. Goremychkin, A. Pozharskii, A. Antonov, P. Tolstoy, A. Filarowski, “Symmetry/asymmetry of NHN hydrogen bond in protonated 1,8-bis(dimethylamino)naphthalene”, Symmetry 2020, 12, 1924. DOI: 10.3390/sym12111924. V. Mikshiev, A.F. Pozharskii, A. Filarowski, A.S. Novikov, A.S. Antonov, P.M. Tolstoy. M.A. Vovk, O.V. Khoroshilova, “How strong is hydrogen bonding to amide nitrogen?”, Chem. Phys. Chem. 2020, 21, 651-658. DOI: 10.1002/cphc.201901104. A.F. Pozharskii, V.A. Ozeryanskii, O.V. Dyablo, O.G. Pogosova, G.S. Borodkin, A. Filarowski, “Nucleophilic Substitution of Hydrogen Atom in Initially Inactivated Pyrrole Ring”, Org. Lett. 2019, 21(7), 1953-1957. DOI: 10.1021/acs.orglett.8b04098. |
|
Dr. Svetlana Pylaeva Chair of Theoretical Chemistry, University of Paderborn, Paderborn, Germany |
M.A. Kostin, S.A. Pylaeva, P.M. Tolstoy, “Phosphine oxides as NMR and IR spectroscopic probes for the estimation of the geometry and energy of hydrogen bonds: PO…H-A hydrogen bonds”, Phys. Chem. Chem. Phys. 2022, 24, 7121-7133. DOI: 10.1039/D1CP05939D. S.A. Pylaeva, H. Elgabarty, D. Sebastiani, P.M. Tolstoy, “Symmetry and dynamics of FHF– anion in vacuum, in CD2Cl2 and in CCl4. Ab initio MD study of fluctuating solvent-solute hydrogen and halogen bonds”, Phys. Chem. Chem. Phys. 2017, 19, 26107-26120. DOI: 10.1039/C7CP04493C. B. Koeppe, S.A. Pylaeva, C. Allolio, D. Sebastiani, E.T.J. Nibbering, G.S. Denisov, H.-H. Limbach, P.M. Tolstoy, “Polar solvent fluctuations drive proton transfer in hydrogen bonded complexes of carboxylic acid with pyridines: NMR, IR and ab initio MD study”, Phys. Chem. Chem. Phys. 2017, 19, 1010-1028. DOI: 10.1039/C6CP06677A. |
|
Dr. Mark Sigalov Department of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva, Israel |
E.Yu. Tupikina, M. Sigalov, I.G. Shenderovich, V.V. Mulloyarova, G.S. Denisov, P.M. Tolstoy, “Correlations of NHN hydrogen bond energy with geometry and 1H NMR chemical shift difference of NH protons for aniline complexes”, J. Chem. Phys. 2019, 150, 114305. DOI: 10.1063/1.5090180. |
|
Dr. Kostas Sotiriadis Institute of Theoretical and Applied Mechanics of the Czech Academy of Sciences, Prague, Czech Republic |
A.Mazur, P. Tolstoy, K. Sotiriadis, “13С, 27Al and 29Si NMR investigation of the hydration kinetics of Portland-limestone cement pastes containing CH3-COO–R+ (R=H or Na)”, Materials 2022, 15, 2004. DOI: 10.3390/ma15062004. K. Sotiriadis, K. Aspiotis, A. Mazur, P. Tolstoy, E. Badogiannis, S. Tsivilis, “Characterization of Old Concrete from a Heritage Structure of Inousses Cluster of Islands”, Lect. Notes Civ. Eng. 2022, 209, 80-89. DOI: 10.1007/978-3-030-90788-4_7. K. Sotiriadis, P. Mácová, A.S. Mazur, A. Viani, P.M. Tolstoy, S. Tsivilis, “Long-term thaumasite sulfate attack on Portland-limestone cement concrete: A multi-technique analytical approach for assessing phase assemblage”, Cement and Concrete Research 2020, 130, 105995. DOI: 10.1016/j.cemconres.2020.105995. |
|
Dr. Viktor Rozentsvet Institute of Ecology of the Volga River Basin, Russian Academy of Sciences, Tol´yatti, Russia |
V.A. Rozentsvet, D.M. Ulyanova, N.A. Sablina, S.V. Kostjuk, P.M. Tolstoy, I.A. Novakov, “Cationic polymerization of butadiene using alkyl aluminum compounds as co-initiators: an efficient approach toward solid polybutadienes”, Polym. Chem. 2022, 3, 1596-1607. DOI: 10.1039/d1py01684a. V. Rozentsvet, N. Sablina, D. Ulyanova, S. Kostjuk, P. Tolstoy, “Structure of terminal units of polybutadiene synthesized via anionic mechanism”, Polym. Bull. 2022, 79, 1239-1256. DOI: 10.1007/s00289-021-03549-5. V.A. Rozentsvet, N.A. Sablina, D.M. Ulyanova, P.M. Tolstoy, I.A. Novakov, “Polymerization of Isoprene Using Cationic Catalytic Systems Based on Triethylaluminum”, Doklady Phys Chem. 2021, 499, 73-76. DOI: 10.1134/S0012501621080017. V.A. Rozentsvet, N.A. Sablina, D.M. Ulyanova, S.N. Smirnov, P.M. Tolstoy, “Structure of the polymer chain of poly(1,3-pentadiene) synthesized using a stereospecific catalytic system”, Russ. Chem. Bull. 2021, 70(4), 773-779. DOI: 10.1007/s11172-021-3150-2. V. Rozentsvet, N. Sablina, D. Ulyanova, S. Kostjuk, P. Tolstoy, “Structure of terminal units of polybutadiene synthesized via anionic mechanism”, Polym. Bull. 2021, accepted. DOI: 10.1007/s00289-021-03549-5. |
|
Dr. Alexander Artem’ev Nikolaev Institute of Inorganic Chemistry, Siberian Branch of Russian Academy of Sciences, Novosibirsk, Russia |
A.Yu. Baranov, A.S. Berezin, D.G. Samsonenko, A.S. Mazur, P.M. Tolstoy, V.F. Plyusnin, I.E. Kolesnikov, A.V. Artem'ev, “New Cu(I) halide complexes showing TADF combined with room temperature phosphorescence: the balance tuned by halogens”, Dalton Trans., 2020, 49, 3155-3163. DOI: 10.1039/D0DT00192A. |
|
Dr. Boris P. Nikolaev Department of Nanomedicine, Research Institute of Highly Pure Biopreparations, St Petersburg, Russia |
M. Shevtsov, B. Nikolaev, Y. Marchenko, L. Yakovleva, N.V. Skvorzov, A. Mazur, P. Tolstoy, V. Ryzhov, G. Multhoff, “Targeting experimental orthotopic glioblastoma with chitosan-based superparamagnetic iron oxide nanoparticles (CS-DX-SPIONs)”, Int. J. Nanomedicine 2018, 13, 1471-1482. DOI: 10.2147/IJN.S152461. |
|
Dr. Irina V. Terekhova G.A. Krestov Institute of Solution Chemistry of Russian Academy of Sciences, Ivanovo, Russia |
I. Kritskiy, T. Volkova, S. Ivanov, A. Mazur, P. Tolstoy, I. Terekhova, “Methotrexate-Loaded Metal-Organic Frameworks on the Basis of γ-Cyclodextrin: Design, Characterization, in Vitro and in Vivo Investigation”, Materials Science and Engineering C 2020, 111, 10774. DOI: 10.1016/j.msec.2020.110774. |
|
Dr. Viacheslav E. Eremyashev South Ural State University, Chelyabinsk, Russia Institute of Mineralogy, Ural Branch, Russian Academy of Sciences, Miass, Russia |
V.E. Eremyashev, A.S. Mazur, P.M. Tolstoi, L.M. Osipova, “Structure of Rubidium Borosilicate Glasses Studied by Nuclear Magnetic Resonance Spectroscopy”, Inorganic Materials 2019, 55, 500-505. DOI: 10.1134/S0020168519050054. |
Research
Non-covalent Interactions
Non-covalent interaction is the common name for interactions the formation of which does not result in the commonization of the electron density. The energy of non-covalent interactions is much lower than the energy of covalent ones. However, due to their prevalence, non-covalent interactions have a significant influence on the physical, chemical, and structural properties of molecules. These mobile and variable interactions while often not determining the constancy of the composition of a chemical compound in different phase states, govern many of its physical, chemical, and structural properties. These interactions can be found in biopolymers, liquid crystals, supramolecular aggregates, solutions, liquids, and molecular crystals. Non-covalent interactions are ubiquitous, numerous, and functional: every chemical reaction starts with them and every condensed state of matter is maintained by them.
All known to date non-covalent interactions can be classified into four groups: electrostatic, Van-der-Waals, hydrophobic, and π-interaction.
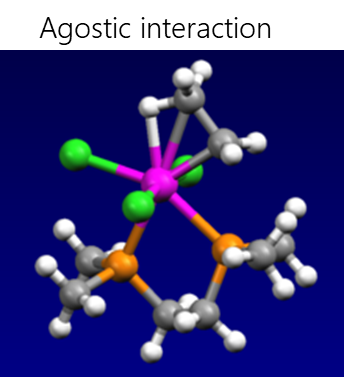
Electrostatic interactions include hydrogen bonding, σ-hole interactions (halogen, pnictogen, halcogen, aerogen, tetrel etc.), agnostic interactions and ionic interactions.

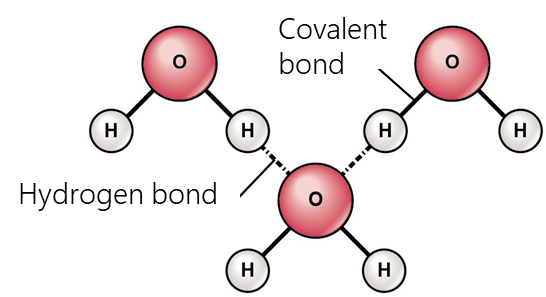
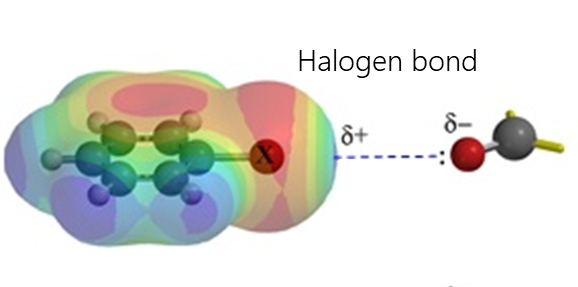
- Halogen bonding occurs due to an attractive interaction between the electrophilic region of the halogen atom (the so-called σ-hole) and the nucleophilic region (such as an unshared electron pair) in another molecule. This interaction is widely used in such fields as supramolecular chemistry, crystallography, and liquid crystal fabrication.
- Hydrogen bonding occurs when a hydrogen atom bonded to a strongly negative atom (fluorine, oxygen, nitrogen, etc.) comes to proximity with another electronegative atom. These bonds are usually stronger than normal dipole-dipole and dispersion forces, but weaker than true covalent and ionic bonds. Hydrogen bonding largely determines the properties of such biologically important substances as proteins and nucleic acids. In particular, secondary structure (e.g., α-helices, β-folds) and tertiary structure in proteins, RNA, and DNA molecules are stabilized by hydrogen bonds.
- Ionic interaction is realized by electrostatic attraction between anions and cations. The most important features of ionic bonding from other types of chemical bonding are its non-directional and non-saturated nature. That is why crystals formed by ionic bonding tend to have various dense packings of the corresponding ions.
- Agostic interaction is the interaction of a coordinationally unsaturated transition metal with the C-H bond, when the two electrons involved in the C-H bond move to the empty d-orbital of the transition metal, resulting in the formation of a three-center two-electron bond. Many catalytic transformations, such as oxidative addition and reductive elimination, presumably involve agostic interactions.
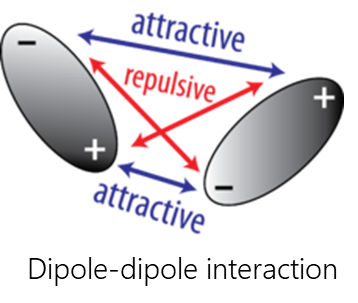
Van-der-Waals interactions include dipole-dipole, dipole-induced dipole, and dispersive interactions.
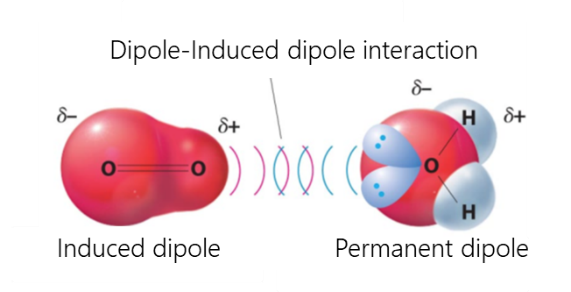
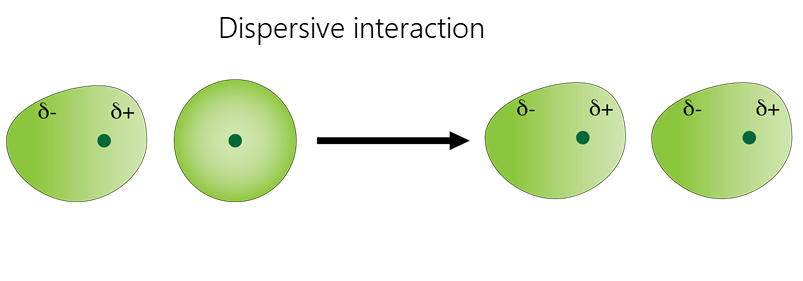
- Dispersive interaction is caused by the interaction of atoms and molecules with each other due to their own or mutually induced dipole moments. The instantaneous charge distribution of one atom or molecule, characterized by an instantaneous dipole moment, induces an instantaneous dipole moment in another atom or molecule
- The interaction between a dipole and an induced dipole occurs when a polar molecule induces a dipole in an atom or in a non-polar molecule, disturbing the arrangement of electrons in the non-polar member.
- Dipole-dipole interactions occur when two polar molecules interact with each other through space. In this case, the partially negative part of one of the polar molecules is attracted to the partially positive part of the second polar molecule.
Hydrophobic interactions are the attraction between non-polar particles in water (or other polar solvents), which is caused by the thermodynamic disadvantage of water contact with non-polar substances. Hydrophobic interaction takes part in the formation of the tertiary structure of proteins and provides molecular recognition in some supramolecular guest-host complexes. It manifests itself in the formation of micelles and other structures in surfactant solutions.

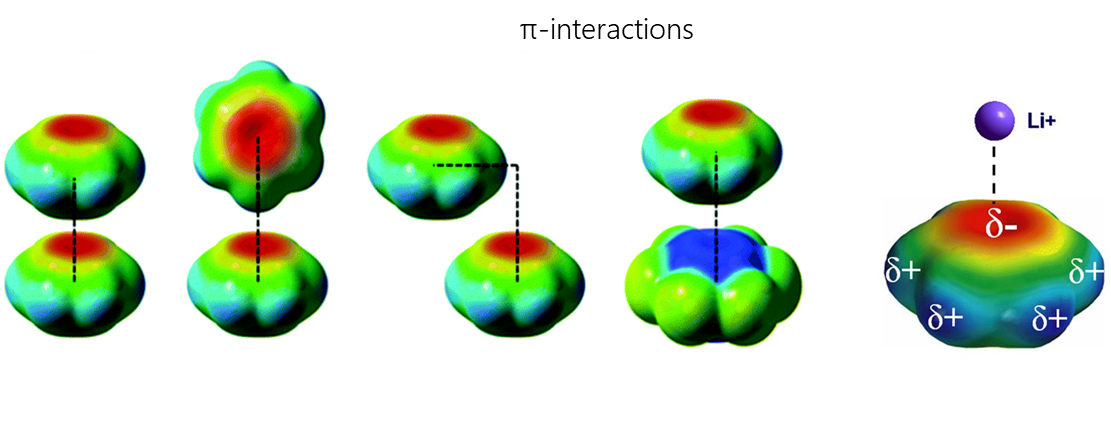
π-interactions are a type of non-covalent interactions in which an electron-rich π-system can interact with a cation, anion or other π-system. Non-covalent interactions with π-systems are crucial for biological processes such as protein-ligand recognition.
Projects
Organometallic Chemistry
Since the discovery of the basic principles of organometallic compounds by Edward Frankland in the second half of the 19th century, the application of these reagents in organic synthesis began to develop rapidly. Today, the organometallic strategy of functionalization makes it possible to carry out complex transformations with high regio- and stereoselectivity and opens the possibility of introducing substituents in positions that are inaccessible by methods of classical chemistry. In order to further expand the synthetic potential of organoelement compounds, the following projects are being implemented in our laboratory:
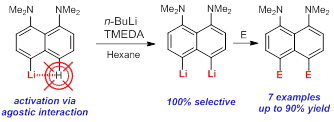
- Direct lithiation of aromatic amines and related compounds for the synthesis of their hard-to-reach meta- and peri-derivatives.
 |
 |
| Antonov, Yakubenko Synthesis 2020 | Antonov et al. J. Organomet. Chem. 2020 |
- Study of the structure of organometallic compounds.
Antonov et al. Organometallics. 2020
- Steric activation of the dimethylamino group in the synthesis of polynuclear nitrogen heterocycles.
Antonov et al. Molecules 2020
Modern approach to the study of non-covalent interactions requires the development of methods for the synthesis of new donors and acceptors, which will allow to understand the periodicity of changes in the interaction force from the nature of the elements that make up the binding participants, as well as allow the use of new more sensitive methods of investigation. Within the framework of this direction our laboratory implements the following projects:
- Development of methods for the synthesis of organic acids, oxides, and selenides of group 15 elements.
Yakubenko et al. Phys. Chem. Chem. Phys. 2022
- Synthesis of chiral selenophosphoric acids and study of their catalytic activity ( in collaboration with the University of Regensburg).
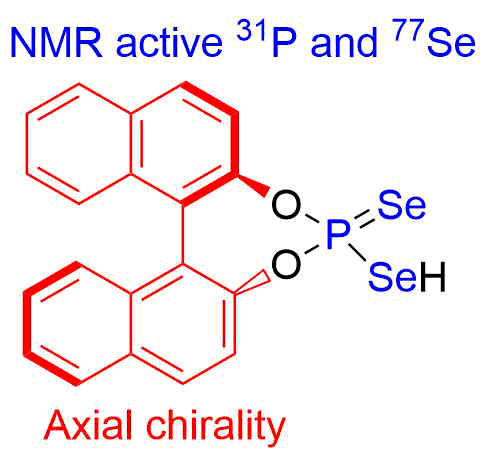
Quantum Chemistry
Quantum chemistry methods are based on the principles of quantum mechanics applied to molecular systems. The solution of the quantum chemical problem is reduced to the numerical solution of the Schrödinger equation using various approximations. Modern methods require significant computational resources, therefore calculations are carried out using powerful computational clusters.
Spectroscopy (IR, NMR) and quantum chemistry methods are complement each other. Calculations are used to explain experimentally observed phenomena, to predict experimental results, or to model impossible experiments.
In our laboratory, we solve the problem of finding correlations between the spectral parameters of complexes with hydrogen bonds and their geometry and energy.
 |
 |
 |
| Tupikina et al. Phys. Chem. Chem. Phys, 2021 | Karpov et al. J. Comput. Chem. 2021 | Tupikina et al. Spectrochim. Acta A 2022 |
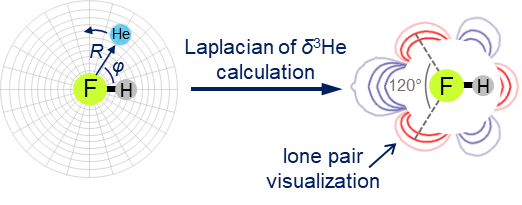
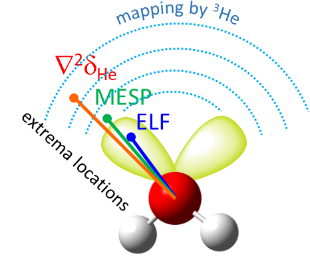
We have proposed a new method for quantum-chemical diagnostics of the features of the outer electronic shell of atoms, molecules and complexes with hydrogen bonds. The keystone of this approach is usage of the 3He atom as a probe particle. Our method allows visualization of both proton-acceptor and proton-donor properties of molecular systems and has advantages over conventional methods (MESP, ELF).
 |
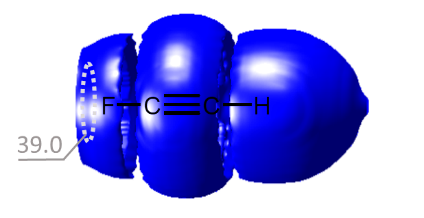 |
 |
| Tupikina et al. J. Phys. Chem. A 2017 | Tupikina et al. J. Comput. Chem. 2018 | Tupikina et al. J. Comput. Chem. 2020 |
One of the directions of quantum-chemical research in our laboratory is the study of the features of the electronic shells of fragments of biological molecules (amino acids, proteins, enzymes). In particular, prediction of possible directions of nucleophilic attack, study of the influence of additional hydrogen bonds with solvent molecules or side chains of a biological molecule on its reactivity.
NMR Spectroscopy
The nuclear magnetic resonance technique (NMR) is based on the registration of resonant absorption of radio frequency energy by a sample placed in a uniform magnetic field. Radiation is absorbed by nuclei with a nonzero magnetic moment (1H, 13C, 19F, 31P, etc.). NMR spectroscopy is the most important method for determining the structure of organic molecules.
In our laboratory, experimental NMR spectroscopy is used to study the properties of various systems with hydrogen and halogen bonds. In particular, we are working on the development of a methodology for interpreting the experimental values of 31P chemical shifts as descriptors of the energy and geometry of the hydrogen bond. Establishing quantitative relationships between the 31P chemical shift is complicated (and therefore becomes more curious :)) due to the fact that the phosphorus nucleus is extremely sensitive to many additional factors.
 |
 |
|
| Mulloyarova et al. J. Molec. Struct. 2021 | Giba et al. Symmetry, 2021 | Yakubenko et al. Phys. Chem. Chem. Phys. 2022 |
To obtain high-resolution NMR spectra at low temperatures (up to 100 K), we use and refine a unique technique – we use mixtures of liquefied freon gases as solvents. Under such conditions, the processes of proton and molecular exchange are slowed down and signals of complexes of various stoichiometric and isotopic compositions are resolved on the NMR spectra.
Mulloyarova et al. Phys. Chem. Chem. Phys. 2018
Our grants
- RFBR grant 20-03-00231 «Media effects on proton position in hydrogen bond: local field and specific solvation» (Dr. P. Tolstoy).
- RSF grant 21-73-10040 «Organolithium reagents in the synthesis and functionalisation of nitrogen heterocycles» (Dr. A. Antonov).
- RSF grant 18-13-00050 «Spectral diagnosis of non-covalent interactions» (Dr. P. Tolstoy).
Publications
Selected publications 2020–2022
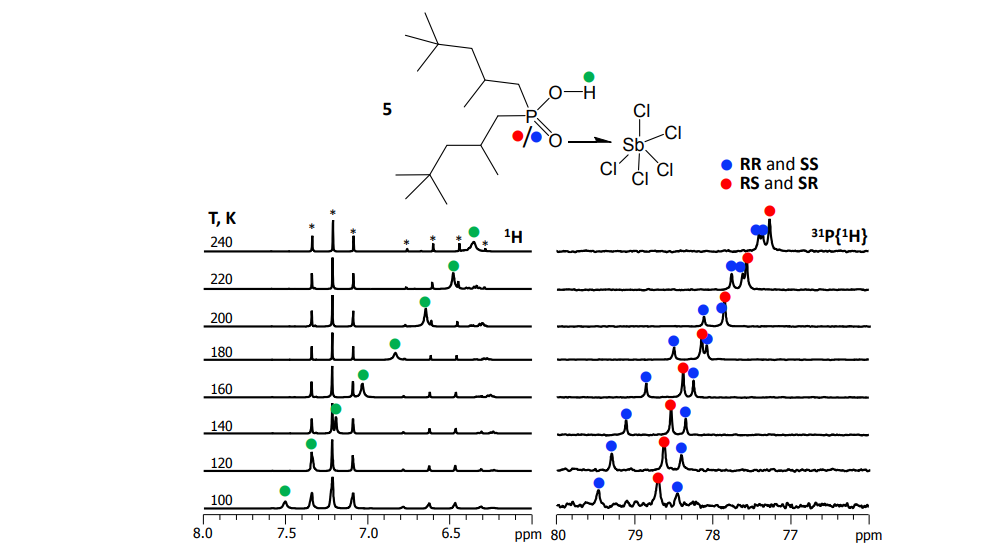
M.A. Kostin, S.A. Pylaeva, P.M. Tolstoy, “Phosphine oxides as NMR and IR spectroscopic probes for the estimation of the geometry and energy of hydrogen bonds: PO…H-A hydrogen bonds”, Phys. Chem. Chem. Phys. 2022, 24, 7121-7133. DOI: 10.1039/D1CP05939D.
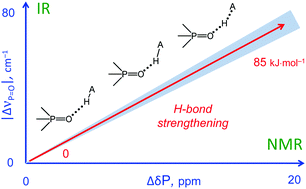 In this work we evaluate the possibility of using the NMR and IR spectral properties of the PO group to estimate the geometry and strength of hydrogen bonds which it forms with OH-, NH- and CH-acids. The results of the DFT study of 70 hydrogen-bonded 1 : 1 complexes of a model trimethylphosphine oxide, Me3PO, with various proton donors in the gas phase and in aprotic medium (modelled as a polarizable continuum) are presented. Four types of hydrogen bonds with the general formula Me3PO⋯H–A were considered, where the A atom is O, C, and N (neutral or cationic acids). Within the selected set of complexes the hydrogen bond energy varies over a wide range (ca. 0–85 kJ mol−1). We show that it is possible to use simple correlations to estimate the energy and geometry of OHO, NHO and CHO hydrogen bonds from the changes of isotropic 31P NMR chemical shifts and harmonic PO stretching vibration frequencies upon complexation. Such correlations also could be used to estimate the proton-donating ability (and Brønsted acidity; pKa) of OH acids.
In this work we evaluate the possibility of using the NMR and IR spectral properties of the PO group to estimate the geometry and strength of hydrogen bonds which it forms with OH-, NH- and CH-acids. The results of the DFT study of 70 hydrogen-bonded 1 : 1 complexes of a model trimethylphosphine oxide, Me3PO, with various proton donors in the gas phase and in aprotic medium (modelled as a polarizable continuum) are presented. Four types of hydrogen bonds with the general formula Me3PO⋯H–A were considered, where the A atom is O, C, and N (neutral or cationic acids). Within the selected set of complexes the hydrogen bond energy varies over a wide range (ca. 0–85 kJ mol−1). We show that it is possible to use simple correlations to estimate the energy and geometry of OHO, NHO and CHO hydrogen bonds from the changes of isotropic 31P NMR chemical shifts and harmonic PO stretching vibration frequencies upon complexation. Such correlations also could be used to estimate the proton-donating ability (and Brønsted acidity; pKa) of OH acids.
E. Tupikina, A.A. Titova, M.V. Kaplanskiy, E.R. Chakalov, M.A. Kostin, P.M. Tolstoy, “Estimations of OH···N hydrogen bond length from positions and intensities of IR bands”, Spectrochimica Acta A 2022, published online. DOI: 10.1016/j.saa.2022.121172
In this computational work applicability of IR spectral parameters for evaluations of OH···N hydrogen bond length is discussed. For a set of 124 complexes with OH···N hydrogen bond formed by combinations of methanol/acetic acid and pyridine (and their fluorine substituted versions) geometries, energies and IR parameters were calculated at MP2/def2-TZVP level of theory. For a number of IR parameters (the shift of proton donor group stretching vibration ∆ns, increase of its intensity I, the low-frequency hydrogen bond stretching vibration nσ, bending in-plane d and out-of-plane g vibrations) equations linking them with interatomic distances are proposed, the robustness and accuracy of such equations are discussed. The enthalpy of OH···N hydrogen bond formation ∆H was also linked with electron density parameters in (3; –1) critical point.
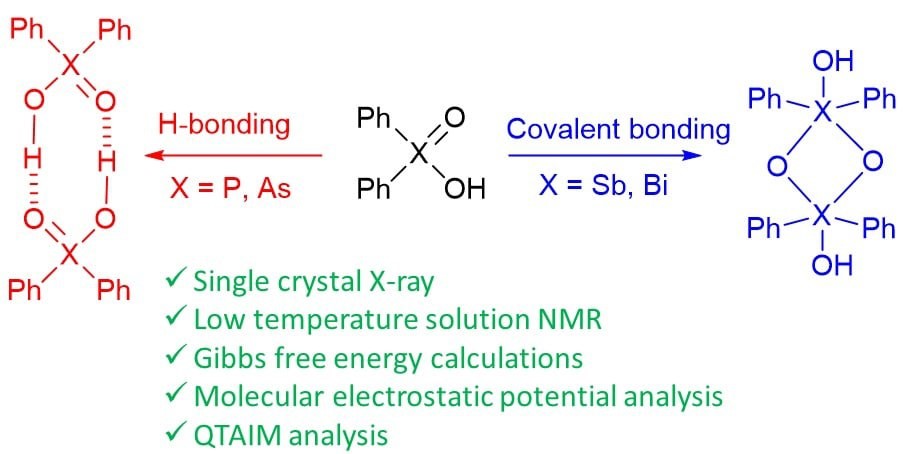
A. Yakubenko, A. Puzyk, V. Korostelev, V. Mulloyarova, E. Tupikina, P. Tolstoy, A. Antonov, “Oxidation of triphenylpnictogens and complexation of resulted di-phenylpnictoginic acids in solution and solid state”, Phys. Chem. Chem. Phys. 2022, accepted. DOI: 10.1039/D2CP00286H.
Triphenylpnictogens were oxidazed to access diphenylpnictiogenic acids Ph2XOOH (X = P, As, Sb, Bi). It was shown that oxidation with chloramine-T does not lead to the cleavage of a C–pnictogen bond. The preliminary reductive cleavage with sodium in liquid ammonia followed by the oxidation with hydrogen peroxide was successfully utilised for the synthesis of diphenylphosphinic and diphenylarsinic acids. It was shown that in solid state (by means of XRD), all diphenylpnictoginic acids form polymeric chains. Diphenylbismuthinic and diphenylantimonic acids form polymeric covalent adducts, while diphenylphosphinic and diphenylarsinic chains are associated through hydrogen bonding. Unlike diphenylphosphinic acid, diphenilarsinic acid forms two polymorphs of hydrogen-bonded infinite chains. In solution in a polar aprotic solvent diphenylarsinic acid, similarly to dimethylarsinic, forms hydrogen-bonded cyclic dimers together with a small amount of cyclic trimers.
A.A. Yakubenko, V.V. Karpov, E.Yu. Tupikina, A.S. Antonov, “Lithiation of 2,4,5,7-Tetrabromo-1,8-bis(dimethylamino)naphthalene: Peculiarities of Directing Groups' Effects and the Possibility of Polymetalation”, Organometallics 2021, 40(21), 3627–36368. DOI: 10.1021/acs.organomet.1c00489.
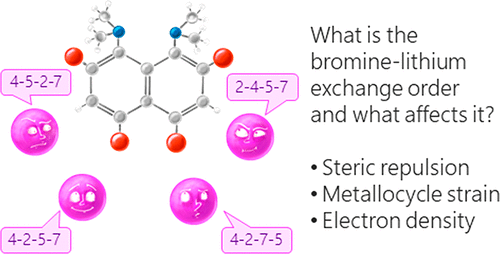 The bromine-lithium exchange sequence upon interaction of 2,4,5,7-tetrabromo-1,8-bis(dimethylamino)naphthalene with n-BuLi was studied. Experimental results were explained by means of quantum chemical calculations. It was demonstrated that the first exchange occurs in position 4 due to the significant decrease of a steric strain of the molecule. The second exchange takes place in either position 5 or 7 due to the even more negative charge distribution in the naphthalene core. As a result, the third exchange leads to the species containing lithium in positions 2,4,5 or 2,4,7. Using a large excess of n-BuLi in hexane, 2,4,5,7-tetralithio-1,8-bis(dimethylamino)naphthalene was successfully prepared. The latter was used for the synthesis of several tetrasubstituted derivatives of 1,8-bis(dimethylamino)naphthalene by quenching with different electrophiles.
The bromine-lithium exchange sequence upon interaction of 2,4,5,7-tetrabromo-1,8-bis(dimethylamino)naphthalene with n-BuLi was studied. Experimental results were explained by means of quantum chemical calculations. It was demonstrated that the first exchange occurs in position 4 due to the significant decrease of a steric strain of the molecule. The second exchange takes place in either position 5 or 7 due to the even more negative charge distribution in the naphthalene core. As a result, the third exchange leads to the species containing lithium in positions 2,4,5 or 2,4,7. Using a large excess of n-BuLi in hexane, 2,4,5,7-tetralithio-1,8-bis(dimethylamino)naphthalene was successfully prepared. The latter was used for the synthesis of several tetrasubstituted derivatives of 1,8-bis(dimethylamino)naphthalene by quenching with different electrophiles.
B. Koeppe, J. Guo, P.M. Tolstoy, H.-H. Limbach, “NMR and UV-vis spectroscopic studies of models for the hydrogen bond system in the active site of photoactive yellow protein: hydrogen bond cooperativity and medium effects”, J. Phys. Chem. B 2021, 125, 22, 5874-5884. DOI: 10.1021/acs.jpcb.0c09923.
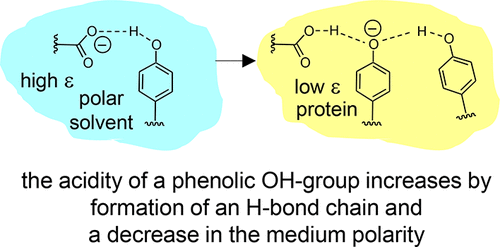 Intramolecular hydrogen bonds in aprotic media were studied by combined (simultaneous) NMR and UV–vis spectroscopy. The species under investigation were anionic and featured single or coupled H-bonds between, for example, carboxylic groups and phenolic oxygen atoms (COO···H···OC)−, among phenolic oxygen atoms (CO···H···OC)−, and hydrogen bond chains between a carboxylic group and two phenolic oxygen atoms (COO···H···(OC)···H···OC)−. The last anion may be regarded as a small molecule model for the hydrogen bond system in the active site of wild-type photoactive yellow protein (PYP) while the others mimic the corresponding H-bonds in site-selective mutants. Results of this work obtained from the model systems to PYP suggests that both cooperative effects within the hydrogen bond chain and a low-polarity protein environment are prerequisites for the stabilization of negative charge on the cofactor and hence for the spectral tuning of the photoreceptor.
Intramolecular hydrogen bonds in aprotic media were studied by combined (simultaneous) NMR and UV–vis spectroscopy. The species under investigation were anionic and featured single or coupled H-bonds between, for example, carboxylic groups and phenolic oxygen atoms (COO···H···OC)−, among phenolic oxygen atoms (CO···H···OC)−, and hydrogen bond chains between a carboxylic group and two phenolic oxygen atoms (COO···H···(OC)···H···OC)−. The last anion may be regarded as a small molecule model for the hydrogen bond system in the active site of wild-type photoactive yellow protein (PYP) while the others mimic the corresponding H-bonds in site-selective mutants. Results of this work obtained from the model systems to PYP suggests that both cooperative effects within the hydrogen bond chain and a low-polarity protein environment are prerequisites for the stabilization of negative charge on the cofactor and hence for the spectral tuning of the photoreceptor.
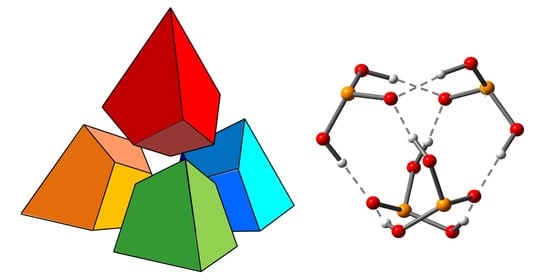
I.S. Giba, P.M. Tolstoy, “Self-assembly of tetrahedral hydrogen-bonded cage tetramers of phosphonic acid”, Symmetry 2021, 13, 258. DOI: 10.3390/sym13020258.
The self-association of phosphonic acids with general formula RP(O)(OH)2 in solution state remains largely unexplored. The general understanding is that such molecules form multiple intermolecular hydrogen bonds, but the stoichiometry of self-associates and the bonding motifs are unclear. In this work, we report the results of the study of self-association of tert-butylphosphonic acid using low temperature liquid-state 1H and 31P NMR spectroscopy (100 K; CDF3/CDF2Cl) and density functional theory (DFT) calculations. For the first time, we demonstrate conclusively that polar aprotic medium tert-butylphosphonic acid forms highly symmetric cage-like tetramers held by eight OHO hydrogen bonds, which makes the complex quite stable. In these associates. each phosphonic acid molecule is bonded to three other molecules by forming two hydrogen bonds as proton donor and two hydrogen bonds as proton acceptor.
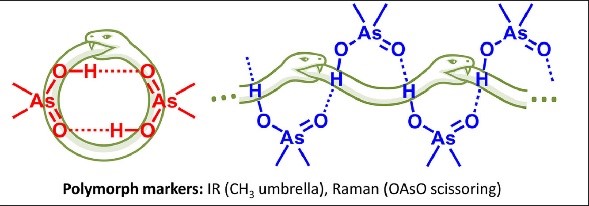
V.V. Mulloyarova, A.M. Puzyk, A.A. Efimova, A.S. Antonov, R.A. Evarestov, I.S. Aliyarova, R.E. Asfin, P.M. Tolstoy, “Solid-State and Solution-State Self-Association of Dimethylarsinic Acid: IR, NMR and Theoretical Study”, J. Mol. Struct. 2021, 1234, 130176. DOI: 10.1016/j.molstruc.2021.130176.
The structure of dimethylarsinic acid (Me2AsOOH, DMA) in crystal and in solution in aprotic medium was studied by X-ray, spectroscopic (IR, NMR and Raman) and quantum chemical calculation (DFT) methods. Two polymorphs of DMA - hydrogen-bonded cyclic dimers or infinite chains - were obtained by growing crystals from protic and aprotic solvents. By comparison of the experimental data with the results of quantum-chemical calculations performed with periodic boundary conditions we propose ways how to distinguish two polymorphs using IR marker bands. The self-association of DMA in polar aprotic solution was studied by low-temperature NMR (T = 100 K, solvent CDF3/CDF2Cl mixture).
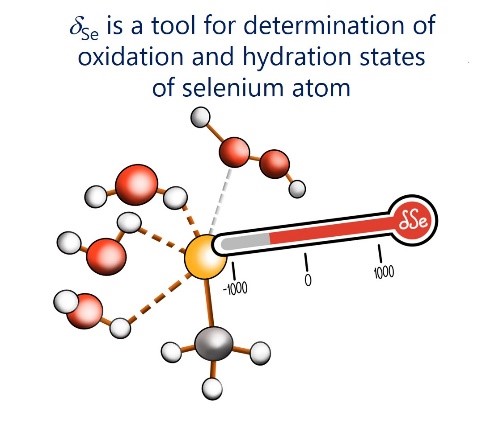
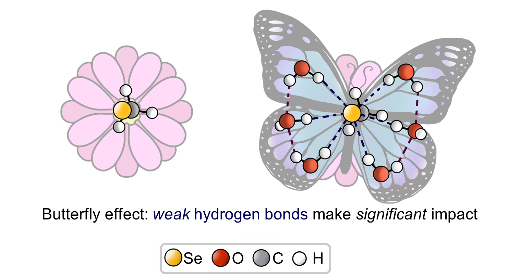
E.Yu. Tupikina, V.V. Karpov, P.M. Tolstoy, “On the influence of water molecules on the outer electronic shells of R–SeH, R–Se(–) and R–SeOH fragments in selenocysteine amino acid residue”, Phys. Chem. Chem. Phys. 2021, 23, 13965-13970. DOI: 10.1039/D1CP01345A.
In this computational work (MP2/aug-cc-pVTZ) we investigated the features of the outer electronic shells of R–SeH, R–Se(−) and R–SeOH fragments (R = CH3), which can be considered as simplified models for the forms of the active centres of glutathione peroxidases GPx along their catalytic pathway (reduction of peroxides). It is shown that the preferential direction of a nucleophilic attack on the R–Se(−) fragment by a peroxide molecule is determined by the presence of the electron-depleted region of the selenium atom in front of the C–Se bond and nucleophilic attack can be facilitated by the solvation of R–Se(−) by water molecules. Such solvation does not block the direction of potential nucleophilic attack and also leads to the increase of the maximal value of the molecular electrostatic potential on the selenium atom. It was shown that the 77Se NMR chemical shift is sensitive both to the oxidation state and the hydration state of the selenium-containing fragment.
V. Karpov, A. Puzyk, P. Tolstoy, E. Tupikina, “Hydration of selenolate moiety: ab initio investigation of properties of O–H···Se(–) hydrogen bonds in CH3Se(–)∙∙∙(H2O)n clusters”, J. Comput. Chem. 2021, 42, 2014-2023. DOI: 10.1002/jcc.26733.
This work is devoted to investigations of the influence of O–H···Se(–) hydrogen bonds on the electronic shells of selenolate R–Se(–) fragment (R═CH3). The geometric, energetic and nuclear magnetic resonance (NMR) spectral parameters for various conformers of CH3Se(–)⋯(H2O)n clusters with n = 0–6 were calculated at CCSD/aug-cc-pVDZ level of theory. For selenolate anion CH3Se(–) solvation free energy was calculated, and for water media it is equal to −71.41 kcal/mol. For O–H···Se(–) hydrogen bonds the proportionality coefficients between QTAIM parameters at (3; −1) bond critical point and the strength of an individual hydrogen bond ∆E were proposed. It was shown, that despite a relative weakness of O–H···Se(–) hydrogen bonds, the outer electronic shell of the selenium atom changes significantly upon formation of each hydrogen bond. This, in turn, cause the dramatic change of NMR parameters of selenium nuclei.
E.Yu. Tupikina, P.M. Tolstoy, A.A. Titova, M.A. Kostin, G.S. Denisov, “Estimations of FH···X hydrogen bond energies from IR intensities: Iogansen's rule revisited”, J. Comput. Chem. 41 (2021) published online. DOI: 10.1002/jcc.26482
 In this work the possibility of using the IR intensity of the stretching vibration νs of proton donor group for estimation of hydrogen bond strength was investigated. For a set of complexes with FH···X (X = F, N, O) hydrogen bonds in the wide range of energies (0.1–49.2 kcal/mol) vibrational frequencies νs and their intensities A were calculated (CCSD at complete basis set limit). The validity of the previously proposed linear proportionality between the intensification of the stretching vibration νs in IR spectra and hydrogen bond enthalpy –ΔH = 12.2·Δ√A (A.V.Iogansen, Spectrochimica Acta A 1999) was examined. It is shown that for a range of similar hydrogen bond types with complexation energies ∆E < 15 kcal/mol the ∆E(Δ√A) function remains similar to that proposed in the Iogansen’s work, while upon strengthening this dependency becomes significantly non-linear. We examined two other parameters (Δ√A/vs and Δ√A·mR) related to IR intensity as descriptors of hydrogen bond strength which are proportional to transition dipole moment matrix element and mass-independent dipole moment derivative. It was found that the dependency ∆E(Δ√A/vs) stays linear in the whole studied range of complexation energies and it can be used for evaluation of ∆E from infrared spectral data with the accuracy about 2 kcal/mol. The mass-independent product Δ√A·mR is an appropriate descriptor for sets of complexes with various hydrogen bond types. Simple equations proposed in this work can be used for estimations of hydrogen bond strength in various systems, where experimental thermodynamic methods or direct calculations are difficult or even impossible.
In this work the possibility of using the IR intensity of the stretching vibration νs of proton donor group for estimation of hydrogen bond strength was investigated. For a set of complexes with FH···X (X = F, N, O) hydrogen bonds in the wide range of energies (0.1–49.2 kcal/mol) vibrational frequencies νs and their intensities A were calculated (CCSD at complete basis set limit). The validity of the previously proposed linear proportionality between the intensification of the stretching vibration νs in IR spectra and hydrogen bond enthalpy –ΔH = 12.2·Δ√A (A.V.Iogansen, Spectrochimica Acta A 1999) was examined. It is shown that for a range of similar hydrogen bond types with complexation energies ∆E < 15 kcal/mol the ∆E(Δ√A) function remains similar to that proposed in the Iogansen’s work, while upon strengthening this dependency becomes significantly non-linear. We examined two other parameters (Δ√A/vs and Δ√A·mR) related to IR intensity as descriptors of hydrogen bond strength which are proportional to transition dipole moment matrix element and mass-independent dipole moment derivative. It was found that the dependency ∆E(Δ√A/vs) stays linear in the whole studied range of complexation energies and it can be used for evaluation of ∆E from infrared spectral data with the accuracy about 2 kcal/mol. The mass-independent product Δ√A·mR is an appropriate descriptor for sets of complexes with various hydrogen bond types. Simple equations proposed in this work can be used for estimations of hydrogen bond strength in various systems, where experimental thermodynamic methods or direct calculations are difficult or even impossible.
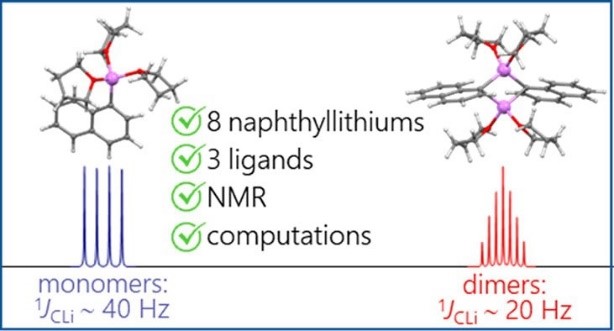
A.S. Antonov, V.V. Karpov, E.Yu. Tupikina, P.M. Tolstoy, M.A. Vovk, “Aggregation Behavior of Lithionaphthalenes in Solution: Experimental and Theoretical Study”, Organometallics 2020, 39, 3705. DOI: 10.1021/acs.organomet.0c00524.
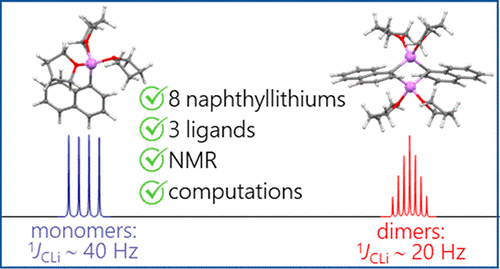 The aggregation of a series of mono- and dilithionaphthalenes in THF solutions in the presence of tetramethylethylenediamine (TMEDA) and pentamethyldiethylenetriamine (PMDTA) was studied by NMR spectroscopy and quantum chemical calculations. The stability of dimeric species in diluted solutions as well as reasons of their disaggregation in some cases has been discussed. For the lithioderivatives of 1,8-bis(dimethylamino)naphthalene (DMAN) the possibility of NMe2 group coordination to the lithium atom in ortho-position of naphthalene core under steric pressure of substituents was demonstrated by quantum chemical calculations. For the first time, dilithionaphthalenes were studied by 1H, 13C, and 7Li NMR spectroscopy. It was shown that while β-dilithionaphthalenes form oligomeric aggregates in solutions, α-dilithionaphthalenes exist as solely monomeric species with each lithium atom coordinated to both peri-positions.
The aggregation of a series of mono- and dilithionaphthalenes in THF solutions in the presence of tetramethylethylenediamine (TMEDA) and pentamethyldiethylenetriamine (PMDTA) was studied by NMR spectroscopy and quantum chemical calculations. The stability of dimeric species in diluted solutions as well as reasons of their disaggregation in some cases has been discussed. For the lithioderivatives of 1,8-bis(dimethylamino)naphthalene (DMAN) the possibility of NMe2 group coordination to the lithium atom in ortho-position of naphthalene core under steric pressure of substituents was demonstrated by quantum chemical calculations. For the first time, dilithionaphthalenes were studied by 1H, 13C, and 7Li NMR spectroscopy. It was shown that while β-dilithionaphthalenes form oligomeric aggregates in solutions, α-dilithionaphthalenes exist as solely monomeric species with each lithium atom coordinated to both peri-positions.
E.Yu. Tupikina, K.G. Tokhadze, V.V. Karpov, G.S. Denisov, P.M. Tolstoy, “Stretching force constants as descriptors of energy and geometry of F···HF hydrogen bonds”, Spectrochim. Acta A 2020, 241, 118677. DOI: 10.1016/j.saa.2020.118677.
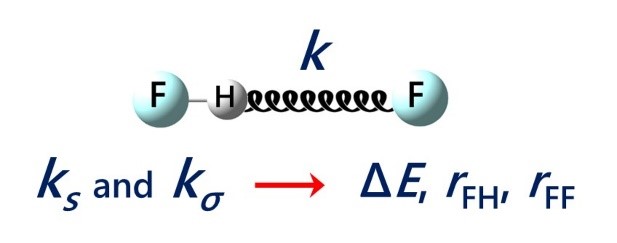 In this work applicability of proton donor group stretching vibration force constants ks and intermolecular stretching force constants kσ for evaluations of hydrogen bond strength and geometry are discussed. For a set of 30 complexes with F···HF hydrogen bonds in a wide range 0.5–48 kcal/mol by means of quantum chemical calculations equilibrium geometries, complexation energies, vibrational frequencies and corresponding force constants were calculated (MP2/aug-cc-pVTZ). It is shown, that properties of a hydrogen bond are more strictly correlated with the values of force constants than with vibrational frequencies. Easy-to-use equations for estimations of hydrogen bond energy ∆E and geometry (rFH, rFF) based on ks and kσ values are proposed.
In this work applicability of proton donor group stretching vibration force constants ks and intermolecular stretching force constants kσ for evaluations of hydrogen bond strength and geometry are discussed. For a set of 30 complexes with F···HF hydrogen bonds in a wide range 0.5–48 kcal/mol by means of quantum chemical calculations equilibrium geometries, complexation energies, vibrational frequencies and corresponding force constants were calculated (MP2/aug-cc-pVTZ). It is shown, that properties of a hydrogen bond are more strictly correlated with the values of force constants than with vibrational frequencies. Easy-to-use equations for estimations of hydrogen bond energy ∆E and geometry (rFH, rFF) based on ks and kσ values are proposed.
A.S. Mazur, M.A. Vovk, P.M. Tolstoy, “Solid-state NMR of carbon nanostructures (graphite, graphene, carbon nanotubes, nanodiamonds, fullerene) in 2000-2019: a mini-review”, Fuller. Nanotub. Car. N. 2020, 28(3), 202-213. DOI: 10.1080/1536383X.2019.1686622.
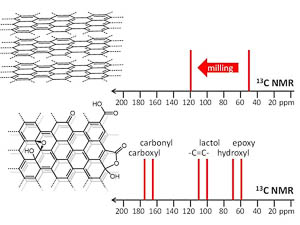 A brief overview is given of the papers published over the last two decades on the subject of solid-state 13C NMR spectra of such carbon nanostructures as milled graphite and its oxide, graphene, graphene oxide, carbon nanotubes, nanodiamonds, fullerenes and their derivatives. The main focus is put on the characteristic values of experimental isotropic 13C NMR chemical shifts and their interpretation in terms of local chemical environments of carbon nuclei.
A brief overview is given of the papers published over the last two decades on the subject of solid-state 13C NMR spectra of such carbon nanostructures as milled graphite and its oxide, graphene, graphene oxide, carbon nanotubes, nanodiamonds, fullerenes and their derivatives. The main focus is put on the characteristic values of experimental isotropic 13C NMR chemical shifts and their interpretation in terms of local chemical environments of carbon nuclei.
V.V. Mulloyarova, D.O. Ustimchuk, A. Filarowski, P.M. Tolstoy, “H/D Isotope Effects on 1H NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids”, Molecules 2020, 25, 1907. DOI: 10.3390/molecules25081907
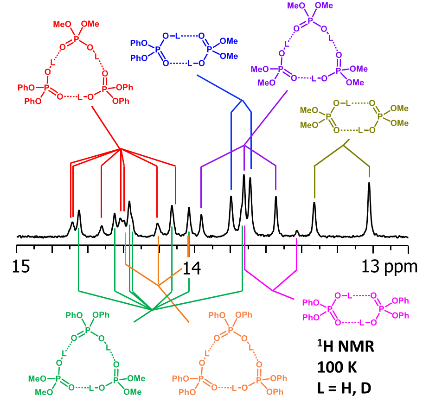 Hydrogen-bonded heterocomplexes formed by POOH-containing acids (diphenylphosphoric 1, dimethylphosphoric 2, diphenylphosphinic 3, and dimethylphosphinic 4) are studied by the low-temperature (100 K) 1H-NMR and 31P-NMR using liquefied gases CDF3/CDF2Cl as a solvent. Formation of cyclic dimers and cyclic trimers consisting of molecules of two different acids is confirmed by the analysis of vicinal H/D isotope effects (changes in the bridging proton chemical shift, δH, after the deuteration of a neighboring H-bond). Acids 1 and 4 (or 1 and 3) form heterotrimers with very strong (short) H-bonds (δH ca. 17 ppm). While in the case of all heterotrimers the H-bonds are cyclically arranged head-to-tail, ···O=P–O–H···O=P–O–H···, and thus their cooperative coupling is expected, the signs of vicinal H/D isotope effects indicate an effective anticooperativity, presumably due to steric factors: when one of the H-bonds is elongated upon deuteration, the structure of the heterotrimer adjusts by shortening the neighboring hydrogen bonds. We also demonstrate the formation of cyclic tetramers: in the case of acids 1 and 4 the structure has alternating molecules of 1 and 4 in the cycle, while in case of acids 1 and 3 the cycle has two molecules of 1 followed by two molecules of 3.
Hydrogen-bonded heterocomplexes formed by POOH-containing acids (diphenylphosphoric 1, dimethylphosphoric 2, diphenylphosphinic 3, and dimethylphosphinic 4) are studied by the low-temperature (100 K) 1H-NMR and 31P-NMR using liquefied gases CDF3/CDF2Cl as a solvent. Formation of cyclic dimers and cyclic trimers consisting of molecules of two different acids is confirmed by the analysis of vicinal H/D isotope effects (changes in the bridging proton chemical shift, δH, after the deuteration of a neighboring H-bond). Acids 1 and 4 (or 1 and 3) form heterotrimers with very strong (short) H-bonds (δH ca. 17 ppm). While in the case of all heterotrimers the H-bonds are cyclically arranged head-to-tail, ···O=P–O–H···O=P–O–H···, and thus their cooperative coupling is expected, the signs of vicinal H/D isotope effects indicate an effective anticooperativity, presumably due to steric factors: when one of the H-bonds is elongated upon deuteration, the structure of the heterotrimer adjusts by shortening the neighboring hydrogen bonds. We also demonstrate the formation of cyclic tetramers: in the case of acids 1 and 4 the structure has alternating molecules of 1 and 4 in the cycle, while in case of acids 1 and 3 the cycle has two molecules of 1 followed by two molecules of 3.
A.S. Ostras’, D.M. Ivanov, A.S. Novikov, P.M. Tolstoy, “Phosphine oxides as spectroscopic halogen bond descriptors: IR and NMR correlations with interatomic distances and complexation energy”, Molecules 2020, 25, 1406. DOI: 10.3390/molecules25061406
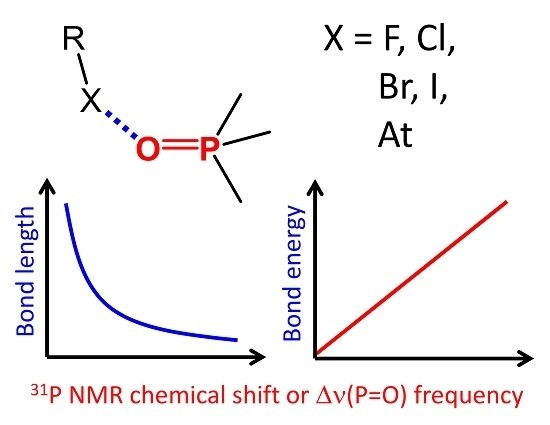 An extensive series of 128 halogen-bonded complexes formed by trimethylphosphine oxide and various F-, Cl-, Br-, I- and At-containing molecules, ranging in energy from 0 to 124 kJ/mol, is studied by DFT calculations in vacuum. The results reveal correlations between R–X···O=PMe3 halogen bond energy ∆E, X···O distance r, halogen’s σ-hole size, QTAIM parameters at halogen bond critical point and changes of spectroscopic parameters of phosphine oxide upon complexation, such as 31P NMR chemical shift, ∆δP, and P=O stretching frequency, ∆ν. Some of the correlations are halogen-specific, i.e., different for F, Cl, Br, I and At, such as ∆E(r), while others are general, i.e., fulfilled for the whole set of complexes at once, such as ∆E(∆δP). The proposed correlations could be used to estimate the halogen bond properties in disordered media (liquids, solutions, polymers, glasses) from the corresponding NMR and IR spectra.
An extensive series of 128 halogen-bonded complexes formed by trimethylphosphine oxide and various F-, Cl-, Br-, I- and At-containing molecules, ranging in energy from 0 to 124 kJ/mol, is studied by DFT calculations in vacuum. The results reveal correlations between R–X···O=PMe3 halogen bond energy ∆E, X···O distance r, halogen’s σ-hole size, QTAIM parameters at halogen bond critical point and changes of spectroscopic parameters of phosphine oxide upon complexation, such as 31P NMR chemical shift, ∆δP, and P=O stretching frequency, ∆ν. Some of the correlations are halogen-specific, i.e., different for F, Cl, Br, I and At, such as ∆E(r), while others are general, i.e., fulfilled for the whole set of complexes at once, such as ∆E(∆δP). The proposed correlations could be used to estimate the halogen bond properties in disordered media (liquids, solutions, polymers, glasses) from the corresponding NMR and IR spectra.
E.Yu. Tupikina, G.S. Denisov, P.M. Tolstoy, “Lone Pairs Mapping by Laplacian of 3He NMR Chemical Shift”, J. Comput. Chem. 2020, 41, 1194-1199. DOI: 10.1002/jcc.26166.
Results of approbation of a new quantum mechanical approach of lone pairs visualization, its optimization and testing on a range of model molecules are presented. The main idea of proposed methodology is using 3He atom as a probe for investigating electronic shells of species with lone pairs. As model objects we consider “classical” examples of hydrogen cyanide, methanimine, ammonia, phosphine, formaldehyde, water and hydrogen sulfide. It is shown, that lone pairs can be visualized by means of 3D maps of Laplacian of 3He chemical shift Ñ2δHe. NMR calculations could be performed using level of theory as low as B3LYP/6-31G, allowing for the reduction of computational time without significant loss of quality. Advantages of our approach are discussed in comparison with usual methods of lone pairs visualization (ELF, MESP).
E. Yu. Tupikina, G. S. Denisov, A. S. Antonov, P. M. Tolstoy, “Unusual behaviour of the spin-spin coupling constant 1JCH upon formation of CH···X hydrogen bond”, Phys. Chem. Chem. Phys. 2020, 22, 1994-2000. DOI: 10.1039/C9CP05964D
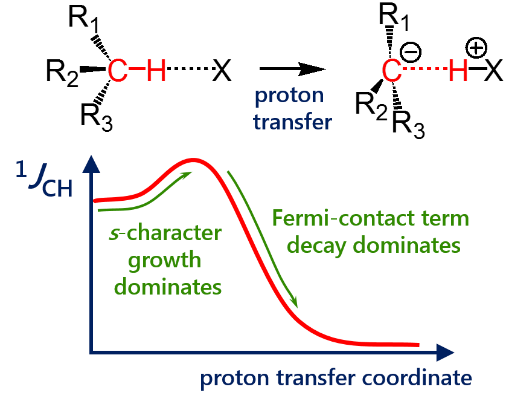 One-bond coupling constants 1JXY are usually used as a measure of corresponding X…Y interatomic distances. However the physical nature of this correlation is not well understood and in some cases a conterintuitive behaviour of 1JXY upon hydrogen bonded complex formation has been reported. In this work the behavior of 1JCH upon formation and strengthening of complexes with CH···X hydrogen bonds and upon proton transfer process is investigated by means of 1H NMR spectroscopy and quantum chemical calculations. 1H NMR spectra of 1,1-dinitroethane solution at room temperature in various solvents (carbon tetrachloride, chloroform, dichloromethane, acetone, dimethylformamide and dimethyl sulfoxide) illustrate the increase of 1JCH by several Hz upon increase of the complex strength. Computational results (MP2/aug-cc-pVDZ) reproduce this observation and allow one to conclude that the increase of 1JCH is mainly caused by the change of the carbon hybridization (an increase of s-character), rather than by the change in interatomic distance rCH. The behavior of 1JCH was also examined computationally in a wide range of CH···X hydrogen bonds’ energies and geometries. For this purpose quantum-chemical modeling of the partial proton transfer process for complexes formed by 1,1-dinitroethane and trinitromethane as hydrogen bond donors with acetone, pyridine and fluoride anion as hydrogen bond acceptors was performed. Obtained results have confirmed the abovementioned idea – for rather weak complexes the dominant impact on the change of 1JCH magnitude is the increase of the s-character of carbon atom hybridization, while for complexes with a significantly transferred proton the exponential decrease of the Fermi-contact term dominates.
One-bond coupling constants 1JXY are usually used as a measure of corresponding X…Y interatomic distances. However the physical nature of this correlation is not well understood and in some cases a conterintuitive behaviour of 1JXY upon hydrogen bonded complex formation has been reported. In this work the behavior of 1JCH upon formation and strengthening of complexes with CH···X hydrogen bonds and upon proton transfer process is investigated by means of 1H NMR spectroscopy and quantum chemical calculations. 1H NMR spectra of 1,1-dinitroethane solution at room temperature in various solvents (carbon tetrachloride, chloroform, dichloromethane, acetone, dimethylformamide and dimethyl sulfoxide) illustrate the increase of 1JCH by several Hz upon increase of the complex strength. Computational results (MP2/aug-cc-pVDZ) reproduce this observation and allow one to conclude that the increase of 1JCH is mainly caused by the change of the carbon hybridization (an increase of s-character), rather than by the change in interatomic distance rCH. The behavior of 1JCH was also examined computationally in a wide range of CH···X hydrogen bonds’ energies and geometries. For this purpose quantum-chemical modeling of the partial proton transfer process for complexes formed by 1,1-dinitroethane and trinitromethane as hydrogen bond donors with acetone, pyridine and fluoride anion as hydrogen bond acceptors was performed. Obtained results have confirmed the abovementioned idea – for rather weak complexes the dominant impact on the change of 1JCH magnitude is the increase of the s-character of carbon atom hybridization, while for complexes with a significantly transferred proton the exponential decrease of the Fermi-contact term dominates.
A.A. Antonov, A.A. Yakubenko, “Noncovalent Li···H Interaction in the Synthesis of peri-Disubstituted Naphthalene Proton Sponges”, Synthesis 2020; 52, 98-104. DOI: 10.1055/s-0039-1690230
Noncovalent Li···H interaction was utilised as a tool for the second lithiation of 4-lithio-1,8-bis(dimethylamino)naphthalene with n-BuLi in the presence of TMEDA in hexane. Metallation proceeds with 100% selectivity in the second peri-position and with up to 90% yield. A series of 4,5-disubstituted derivatives of 1,8-bis(dimethylamino)naphthalene has been prepared in good to excellent yield after quenching the reaction mass with different electrophiles.
V. Mikshiev, A.F. Pozharskii, A. Filarowski, A.S. Novikov, A.S. Antonov, P.M. Tolstoy. M.A. Vovk, O.V. Khoroshilova, “How strong is hydrogen bonding to amide nitrogen?”, Chem. Phys. Chem. 2020, 21, 651-658. DOI: 10.1002/cphc.201901104.
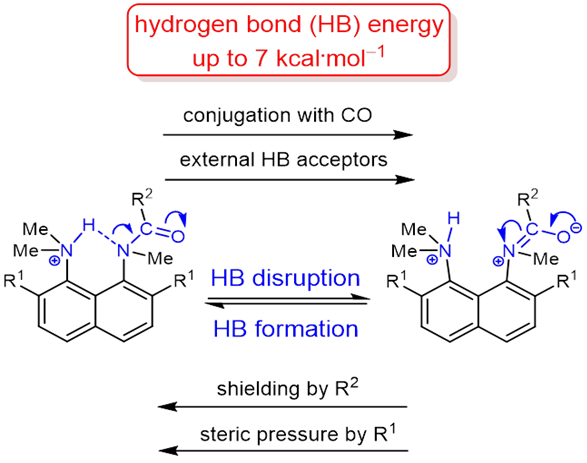 The protonation of the carboxamide nitrogen atom is an essential part of in vivo and in vitro processes (cis‐trans isomerization, amides hydrolysis etc). This phenomenon is well studied in geometrically strongly distorted amides, although there is little data concerning the protonation of undistorted amides. In the latter case, the participation of amide nitrogen in hydrogen bonding (which can be regarded as the incipient state of a proton transfer process) is less well‐studied. Thus, it would be a worthy goal to investigate the enthalpy of this interaction. We prepared and investigated a set of peri‐substituted naphthalenes containing the protonated dimethylamino group next to the amide nitrogen atom (“amide proton sponges”), which could serve as models for the study of an intramolecular hydrogen bond with the amide nitrogen atom. X‐Ray analysis, NMR spectra, basicity values as well as quantum chemical calculations revealed the existence of a hydrogen bond with the amide nitrogen, that should be attributed to the borderline between moderate and weak intramolecular hydrogen bonds (2–7 kcal·mol‒1).
The protonation of the carboxamide nitrogen atom is an essential part of in vivo and in vitro processes (cis‐trans isomerization, amides hydrolysis etc). This phenomenon is well studied in geometrically strongly distorted amides, although there is little data concerning the protonation of undistorted amides. In the latter case, the participation of amide nitrogen in hydrogen bonding (which can be regarded as the incipient state of a proton transfer process) is less well‐studied. Thus, it would be a worthy goal to investigate the enthalpy of this interaction. We prepared and investigated a set of peri‐substituted naphthalenes containing the protonated dimethylamino group next to the amide nitrogen atom (“amide proton sponges”), which could serve as models for the study of an intramolecular hydrogen bond with the amide nitrogen atom. X‐Ray analysis, NMR spectra, basicity values as well as quantum chemical calculations revealed the existence of a hydrogen bond with the amide nitrogen, that should be attributed to the borderline between moderate and weak intramolecular hydrogen bonds (2–7 kcal·mol‒1).
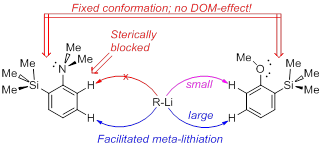
A.S. Antonov, V.G. Bardakov, V.V. Mulloyarova, “Sterically facilitated meta-lithiation of arenes, containing electron-donating groups”, J. Org. Chem. 2020, 906, 121068. DOI: 10.1016/j.jorganchem.2019.121068
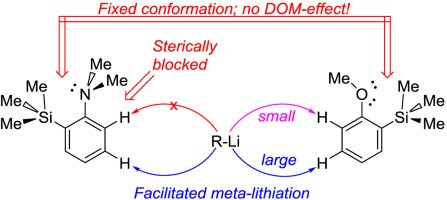 The influence of the bulky trimethylsilyl substituent on the selectivity of metallation of dimethylaniline, anisole and 1-dimethylaminonaphthalene is studied. The neighboring SiMe3 group forces dimethylamino and methoxy groups to occupy a conformation with an unshared electron pair turned towards silicon atom. This forced conformation prevents NMe2 and OMe groups from providing the DOM-effect, thus facilitating meta-metallation with the use of bulky LiCKOR. While the inverted NMe2 group completely suppresses ortho-metallation, the less bulky and more electron withdrawing OMe group demonstrates more rotation freedom allowing selective ortho-metallation with smaller reagents such as n-BuLi or tert-BuLi.
The influence of the bulky trimethylsilyl substituent on the selectivity of metallation of dimethylaniline, anisole and 1-dimethylaminonaphthalene is studied. The neighboring SiMe3 group forces dimethylamino and methoxy groups to occupy a conformation with an unshared electron pair turned towards silicon atom. This forced conformation prevents NMe2 and OMe groups from providing the DOM-effect, thus facilitating meta-metallation with the use of bulky LiCKOR. While the inverted NMe2 group completely suppresses ortho-metallation, the less bulky and more electron withdrawing OMe group demonstrates more rotation freedom allowing selective ortho-metallation with smaller reagents such as n-BuLi or tert-BuLi.
2022
- A. Yakubenko, A. Puzyk, V. Korostelev, V. Mulloyarova, E. Tupikina, P. Tolstoy, A. Antonov, “Oxidation of triphenylpnictogens and complexation of resulted di-phenylpnictoginic acids in solution and solid state”, Phys. Chem. Chem. Phys. 2022, accepted. DOI: 10.1039/D2CP00286H.
- E. Tupikina, A.A. Titova, M.V. Kaplanskiy, E.R. Chakalov, M.A. Kostin, P.M. Tolstoy, “Estimations of OH···N hydrogen bond length from positions and intensities of IR bands”, Spectrochimica Acta A 2022, accepted. DOI: 10.1016/j.saa.2022.121172
- M.A. Kostin, S.A. Pylaeva, P.M. Tolstoy, “Phosphine oxides as NMR and IR spectroscopic probes for the estimation of the geometry and energy of hydrogen bonds: PO…H-A hydrogen bonds”, Phys. Chem. Chem. Phys. 2022, 24, 7121-7133. DOI: 10.1039/D1CP05939D.
- Aliyarova, I.S., Tupikina, E.Yu., Ivanov, D.M., Kukushkin, V. Yu., “Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)Halomethane Cocrystals”, Inorganic Chemistry 2022, 61, 5, 2558–2567. DOI: 10.1021/acs.inorgchem.1c03482.
- K. Sotiriadis, K. Aspiotis, A. Mazur, P. Tolstoy, E. Badogiannis, S. Tsivilis, “Characterization of Old Concrete from a Heritage Structure of Inousses Cluster of Islands”, Lect. Notes Civ. Eng. 2022, 209, 80-89. DOI: 10.1007/978-3-030-90788-4_7.
- M. Nikishina, L. Perelomov, Y. Atroshchenko, E. Ivanova, L. Mukhtorov, P. Tolstoy, “Sorption of fulvic acids and their compounds with heavy metal ions on clay minerals”, Soil Syst. 2022, 6, 2. DOI: 10.3390/soilsystems6010002.
- V. Rozentsvet, N. Sablina, D. Ulyanova, S. Kostjuk, P. Tolstoy, “Structure of terminal units of polybutadiene synthesized via anionic mechanism”, Polym. Bull. 2022, 79, 1239-1256. DOI: 10.1007/s00289-021-03549-5.
- F. Shakirova, U. Markova, P. Tolstoy, A. Bulatov, A. Shishov, “A new hydrophobic deep eutectic solvent based on thymol and 4-(dimethylamino)benzaldehyde: derivatization and microextraction of urea”, J. Mol. Liquids 2022, 353, 118820. DOI: 10.1016/j.molliq.2022.118820.
- A. Mazur, P. Tolstoy, K. Sotiriadis, “13С, 27Al and 29Si NMR investigation of the hydration kinetics of Portland-limestone cement pastes containing CH3-COO–R+ (R=H or Na)”, Materials 2022, 15, 2004. DOI: 10.3390/ma15062004.
- В.А. Розенцвет, Д.М. Ульянова, Н.А. Саблина, М.Г. Кузнецова, П.М. Толстой, «Полимеризация 1,3-пентадиена под действием катионных каталитических систем на основе алюминийорганических соединений», Известия АН 2022, 4, 787-795.
- V.A. Rozentsvet, D.M. Ulyanova, N.A. Sablina, S.V. Kostjuk, P.M. Tolstoy, I.A. Novakov, “Cationic polymerization of butadiene using alkyl aluminum compounds as co-initiators: an efficient approach toward solid polybutadienes”, Polym. Chem. 2022, 3, 1596-1607. DOI: 10.1039/d1py01684a.
2021
- A.A. Yakubenko, V.V. Karpov, E.Yu. Tupikina, A.S. Antonov, “Lithiation of 2,4,5,7-Tetrabromo-1,8-bis(dimethylamino)naphthalene: Peculiarities of Directing Groups' Effects and the Possibility of Polymetalation”, Organometallics 2021, 40(21), 3627–36368. DOI: 10.1021/acs.organomet.1c00489.
- V. Lyutoev, T. Shumilova, A. Mazur, P. Tolstoy, “NMR spectral characteristics of ultrahigh-pressure high-temperature impact glasses of the giant Kara astrobleme (Pay-Khoy, Russia)”, Minerals 2021, 11, 1418. DOI: 10.3390/min11121418.
- В.А. Розенцвет, Н.А. Саблина, Д.М. Ульянова, П.М. Толстой, И.А. Новаков, «Полимеризация изопрена под действием катионных каталитических систем на основе триэтилалюминия», Доклады РАН 2021, 499, 66-70. DOI: 10.31857/S2686953521040063. V.A. Rozentsvet, N.A. Sablina, D.M. Ulyanova, P.M. Tolstoy, I.A. Novakov, “Polymerization of Isoprene Using Cationic Catalytic Systems Based on Triethylaluminum”, Doklady Phys Chem. 2021, 499, 73-76. DOI: 10.1134/S0012501621080017.
- E.Yu. Tupikina, P.M. Tolstoy, A.A. Titova, M.A. Kostin, G.S. Denisov, “Estimations of FH···X hydrogen bond energies from IR intensities: Iogansen's rule revisited”, J. Comput. Chem. 2021, 41. DOI: 10.1002/jcc.26482
- E.Y. Tupikina, S.G. Yastrebov, , “Molecular Complexes of Glycine with Cations H+, Ca2+, and Phosphine Oxide H3PO”, Technical Physics Letters 2021, 47(2), 147–149. DOI: 10.1134/S1063785021020140.
- I.S. Giba, P.M. Tolstoy, “Self-assembly of tetrahedral hydrogen-bonded cage tetramers of phosphonic acid”, Symmetry 2021, 13, 258. DOI: 10.3390/sym13020258.
- I.S. Giba, V.V. Mulloyarova, G.S. Denisov, P.M. Tolstoy, “Sensitivity of 31P NMR chemical shifts to hydrogen bond geometry and molecular conformation for complexes of phosphinic acids with pyridines”, Magn. Reson. Chem. 2021, 59(4), 465–477. DOI: 10.1002/mrc.5123.
- V.V. Mulloyarova, A.M. Puzyk, A.A. Efimova, A.S. Antonov, R.A. Evarestov, I.S. Aliyarova, R.E. Asfin, P.M. Tolstoy, “Solid-State and Solution-State Self-Association of Dimethylarsinic Acid: IR, NMR and Theoretical Study”, J. Mol. Struct. 2021, 1234, 130176. DOI: 10.1016/j.molstruc.2021.130176.
- В.А. Розенцвет, Н.А. Саблина, Д.М. Ульянова, С.Н. Смирнов, П.М. Толстой, «Строение полимерной цепи поли-1,3-пентадиена, синтезированного на стереоспецифической каталитической системе», Известия АН 2021, 4, 773-779. V.A. Rozentsvet, N.A. Sablina, D.M. Ulyanova, S.N. Smirnov, P.M. Tolstoy, “Structure of the polymer chain of poly(1,3-pentadiene) synthesized using a stereospecific catalytic system”, Russ. Chem. Bull. 2021, 70(4), 773-779. DOI: 10.1007/s11172-021-3150-2.
- B. Koeppe, J. Guo, P.M. Tolstoy, H.-H. Limbach, “NMR and UV-vis spectroscopic studies of models for the hydrogen bond system in the active site of photoactive yellow protein: hydrogen bond cooperativity and medium effects”, J. Phys. Chem. B 2021, 125, 22, 5874-5884. DOI: 10.1021/acs.jpcb.0c09923.
- Ł. Hetmańczyk, P. Szklarz, A. Kwocz, M. Wierzejewska, M. Pagacz-Kostrzewa, M.Ya. Melnikov, P.M. Tolstoy, A. Filarowski, “Polymorphism and Conformational Equilibrium for Nitro-Acetophenone in the Solid State and Under the Matrix Condition”, Molecules 2021, 26, 3109. DOI: 10.3390/molecules26113109.
- E.Yu. Tupikina, V.V. Karpov, P.M. Tolstoy, “On the influence of water molecules on the outer electronic shells of R–SeH, R–Se(–) and R–SeOH fragments in selenocysteine amino acid residue”, Phys. Chem. Chem. Phys. 2021, 23, 13965-13970. DOI: 10.1039/D1CP01345A.
- J. Savosina, M. Agafonova-Moroz, M. Khaydukova, A. Legin, V. Babain, P. Tolstoy, D. Kirsanov, “On the radiolytic stability of potentiometric sensors with plasticized polymeric membranes”, Chemosensors 2021, 9, 214. DOI: 10.3390/chemosensors9080214.
- E. Bulatov, T. Eskelinen, A. Ivanov, P. Tolstoy, E. Kalenius, P. Hirva, M. Haukka, “Noncovalent axial I∙∙∙Pt∙∙∙I interactions in platinum(II) complexes strengthen in the excited state”, ChemPhysChem 2021, 22, 1-7. DOI: 10.1002/cphc.202100468.
- Ł. Hetmańczyk, E.A. Goremychkin, J. Waliszewski, M.V. Vener, P. Lipkowski, P.M. Tolstoy, A. Filarowski, “Spectroscopic identification of hydrogen bond vibrations and quasi-isostructural polymorphism in N-salicylideneaniline”, Molecules 2021, 26, 5043. DOI: 10.3390/molecules26165043.
- V. Karpov, A. Puzyk, P. Tolstoy, E. Tupikina, “Hydration of selenolate moiety: ab initio investigation of properties of O–H···Se(–) hydrogen bonds in CH3Se(–)∙∙∙(H2O)n clusters”, J. Comput. Chem. 2021, 42, 2014-2023. DOI: 10.1002/jcc.26733.
- V. Rozentsvet, N. Sablina, D. Ulyanova, S. Kostjuk, P. Tolstoy, “Structure of terminal units of polybutadiene synthesized via anionic mechanism”, Polym. Bull. 2021, accepted. DOI: 10.1007/s00289-021-03549-5.
2020
- K. Sotiriadis, P. Mácová, A.S. Mazur, A. Viani, P.M. Tolstoy, S. Tsivilis, “Long-term thaumasite sulfate attack on Portland-limestone cement concrete: A multi-technique analytical approach for assessing phase assemblage”, Cement and Concrete Research 2020, 130, 105995. DOI: 10.1016/j.cemconres.2020.105995.
- N.M. Kuznetsov, S.I. Belousov, A.V. Bakirov, S.N. Chvalun, R.A. Kamyshinsky, A.A. Mikhutkin, A.L. Vasiliev, P.M. Tolstoy, A.S. Mazur, E.D. Eidelman, E.B. Yudina, A.Ya. Vul', “Unique rheological behavior of detonation nanodiamond hydrosols: the nature of sol-gel transition”, Carbon 2020, 161, 486-494. DOI: 10.1016/j.carbon.2020.01.054.
- A.Yu. Baranov, A.S. Berezin, D.G. Samsonenko, A.S. Mazur, P.M. Tolstoy, V.F. Plyusnin, I.E. Kolesnikov, A.V. Artem'ev, “New Cu(I) halide complexes showing TADF combined with room temperature phosphorescence: the balance tuned by halogens”, Dalton Trans. 2020, 49, 3155-3163. DOI: 10.1039/D0DT00192A.
- I. Kritskiy, T. Volkova, S. Ivanov, A. Mazur, P. Tolstoy, I. Terekhova, “Methotrexate-Loaded Metal-Organic Frameworks on the Basis of γ-Cyclodextrin: Design, Characterization, in Vitro and in Vivo Investigation”, Materials Science and Engineering C 2020, 111, 10774. DOI: 10.1016/j.msec.2020.110774.
- V.А. Rozentsvet, N.А. Sablina, D.M. Ulyanova, P.M. Tolstoy, S.N. Smirnov, I.A. Novakov, “Identification of the structure of polybutadiene terminal units by NMR spectroscopy with T2-filter”, Doklady Phys. Chem. 2020, 491, 40-42. DOI: 10.1134/S0012501620040028.
- V. Rozentsvet, N. Sablina, D. Ulyanova, M. Kuznetsova, S. Kostjuk, P. Tolstoy, “A new approach towards investigation of structure of terminal units of poly(1,3-pentadiene) using T2-filtered NMR spectroscopy”, Macromol. Chem. Phys. 2020, 2000168. DOI: 10.1002/macp.202000168.
- A.S. Antonov, V.V. Karpov, E.Yu. Tupikina, P.M. Tolstoy, M.A. Vovk, “Aggregation Behavior of Lithionaphthalenes in Solution: Experimental and Theoretical Study”, Organometallics 2020, 39, 3705. DOI: 10.1021/acs.organomet.0c00524.
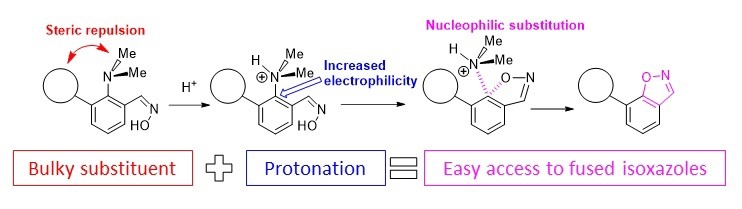
- A.S. Antonov, E.Yu. Tupikina, V.V. Karpov, V.V. Mulloyarova, V.G. Bardakov, “Sterically Facilitated Intramolecular Nucleophilic NMe2 Group Substitution in the Synthesis of Fused Isoxazoles: Theoretical Study”, Molecules, 2020, 25, 5977. DOI: 10.3390/molecules25245977.
- A.S. Antonov, V.G. Bardakov, V.V. Mulloyarova, “Sterically facilitated meta-lithiation of arenes, containing electron-donating groups”, J. Org. Chem. 2020, 906, 121068. DOI: 10.1016/j.jorganchem.2019.121068
- A.A. Antonov, A.A. Yakubenko, “Noncovalent Li···H Interaction in the Synthesis of peri-Disubstituted Naphthalene Proton Sponges”, Synthesis 2020; 52, 98-104. DOI: 10.1055/s-0039-1690230
- E. Yu. Tupikina, G. S. Denisov, A. S. Antonov, P. M. Tolstoy, “Unusual behaviour of the spin-spin coupling constant 1JCH upon formation of CH•••X hydrogen bond”, Phys. Chem. Chem. Phys. 2020, 22, 1994-2000. DOI: 10.1039/C9CP05964D
- E. Yu. Tupikina, K. G. Tokhadze, G. S. Denisov, P. M. Tolstoy, “Lone pairs mapping by Laplacian of 3He NMR chemical shift”, J. Comput. Chem. 2020, 41, 1194-1199. DOI: 10.1002/jcc.26166.
- E.Yu. Tupikina, K.G. Tokhadze, V.V. Karpov, G.S. Denisov, P.M. Tolstoy, “Stretching force constants as descriptors of energy and geometry of F···HF hydrogen bonds”, Spectrochim. Acta A 2020, 241, 118677. DOI: 10.1016/j.saa.2020.118677.
- A.S. Mazur, M.A. Vovk, P.M. Tolstoy, “Solid-state NMR of carbon nanostructures (graphite, graphene, carbon nanotubes, nanodiamonds, fullerene) in 2000-2019: a mini-review”, Fuller. Nanotub. Car. N. 2020, 28(3), 202-213. DOI: 10.1080/1536383X.2019.1686622.
- A.S. Ostras’, D.M. Ivanov, A.S. Novikov, P.M. Tolstoy, “Phosphine oxides as spectroscopic halogen bond descriptors: IR and NMR correlations with interatomic distances and complexation energy”, Molecules 2020, 25, 1406. DOI: 10.3390/molecules25061406.
- V.Y. Mikshiev, A.F. Pozharskii, A. Filarowski, A.S. Novikov, A.S. Antonov, P.M. Tolstoy, M.A. Vovk, O.V. Khoroshilova, “ How strong hydrogen bond with amide nitrogen atom is?”, ChemPhysChem, 2020, 21, 651-658. DOI: 10.1002/cphc.201901104.
- V.V. Mulloyarova, D.O. Ustimchuk, A. Filarowski, P.M. Tolstoy, «H/D Isotope Effects on 1H NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids», Molecules 2020, 25, 1907. DOI: 10.3390/molecules25081907.
- K. Jóźwiak, A. Jezierska, J.J. Panek, E.A. Goremychkin, P.M. Tolstoy, I.G. Shenderovich, A. Filarowski, “Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates”, Molecules 2020, 25, 4720. DOI: 10.3390/molecules25204720.
- V. Torres-Barthelemy, N. Pérez-Hernández, I.G. Shenderovich, P.M. Tolstoy, G.S. Denisov, H.-H. Limbach, “NMR-detected Host-Guest Proton Exchange as a novel Tool to study Surface/Volume Ratios and Filling of Cavities of mesoporous Solids”, J. Phys. Chem. C 2020, 124(40), 22082-22095. DOI: 10.1021/acs.jpcc.0c04889.
- P. Piękoś, A. Jezierska, J. Panek, E.A. Goremychkin, A. Pozharskii, A. Antonov, P. Tolstoy, A. Filarowski, “Symmetry/asymmetry of NHN hydrogen bond in protonated 1,8-bis(dimethylamino)naphthalene”, Symmetry 2020, 12, 1924. DOI: 10.3390/sym12111924.
- S.N. Yunusova, D.S. Bolotin, M.A. Vovk, P.M. Tolstoy, V.Yu. Kukushkin, “Tetrabromomethane as an Organic Catalyst: a Kinetic Study of CBr4-Catalyzed Schiff Condensation”, Eur. J. Org. Chem. 2020, 6763-6769. DOI: 10.1002/ejoc.202001180.
- D.L. Melnikova, Z.F. Badrieva, M.A. Kostin, C. Maller, M. Stas, A. Buczek, M.A. Broda, T. Kupka, A.-M. Kelterer, P.M. Tolstoy, V.D. Skirda, “On Complex Formation Between 5-Fluorouracil and beta-Cyclodextrin in Solution and in the Solid State: IR Markers and Detection of Short-Lived Complexes by Diffusion NMR”, Molecules 2020, 25, 5706. DOI: 10.3390/molecules25235706.
Information for students
We invite bachelor students and 1st year master students to perform investigations for their qualification works in our laboratory in the following areas:
- NMR spectroscopy (Peter M. Tolstoy, room 2150, This email address is being protected from spambots. You need JavaScript enabled to view it.)
- Organometallic chemistry (Alexander S. Antonov, room 2123, This email address is being protected from spambots. You need JavaScript enabled to view it.)
- Quantum chemistry (Elena Yu. Tupikina, room 2123, This email address is being protected from spambots. You need JavaScript enabled to view it.)