First-principles calculations of phonon spectra and thermodynamic properties of transition metal sulfide-based nanotubes
Grant of the Russian Foundation for Basic Research (No. 17-03-00130-a)
Principal investigator: Doctor of Physics and Mathematics, Professor Robert A. Evarestov.
Executors: A. V. Bandura, S. I. Lukyanov, V. V. Porsev, A. V. Kovalenko, D. D. Kuruch
The aim of this project is the development and application of ab initio quantum chemistry methods for calculating thermodynamic properties of nanotubes based on layered transition metal sulfides of 5-th and 6-th periods. Selection of these substances is due to the fact that nanostructures synthesized for transition metal sulfides (TMS) have a great range of properties that can be used in different fields of engineering and technology, such as tribology, catalysis, optoelectronics, and manufacture of batteries, ultracapacitors, and thermoelectric devises. Despite the similarity of their atomic structure, TMS demonstrate a wide range of electronic properties. For example, crystalline zirconium, hafnium, molybdenum and tungsten disulfides are semiconductors; titanium disulfide is semi-metal; whereas niobium and tantalum disulfides are metals. Nanoscale TMS systems have properties that are different from those in the crystalline state. Choosing the composition, morphology, and size of TMS nanosystems, one can strengthen the selected useful properties.
In the first phase of this project we will perform the theoretical analysis of symmetry of phonons and their activity in the IR and Raman spectra of nanotubes based on 3-plane layers with composition MS2 (M = Zr, Hf, Mo, W). Choosing of the metals of 4th and 6th groups is due to the fact that the stable phases of layered sulfides have the different monolayer structure. All considered monolayers are composed of three atomic planes with central plane formed by metal atoms and two outer planes formed by sulfur atoms. However, zirconium and hafnium atoms possess the octahedral coordination, while molybdenum and tungsten atoms have the trigonal prismatic coordination (see fig. 1). Accordingly, we will perform the analysis of the vibrational states of hexagonal layers, resulting from different space groups P-3m (for 1T polymorphic modifications in case of Zr and Hf) and P63/mmc (for 2H polymorphic modification in the case of Mo and W). Consequently, the dynamical representations of corresponding layer groups will be obtained and the symmetry of phonons active IR and Raman spectra of layers will be compared with those for bulk crystals. Diperiodic symmetry group of layers will be used to determine the families of monoperiodic symmetry groups relevant to single-walled nanotubes of different chirality (fig. 2) and their dynamical representations, which will allow us to determine the active IR and Raman spectra of nanotubes in comparison with those for layers. These data are necessary to predict and interpret experimental vibrational spectra of nanotubes considered.
The main goal of our study is to perform the first-principles calculations of phonon spectra of the layers and nanotubes mentioned above. Phonon spectra of layers and nanotubes will be calculated at various points in the Brillouin zone (BZ) to obtain the dispersion curves of phonon states. One of the important objectives of the project is to study the differences between the vibrational spectra of tubes and layers. Quantum chemical calculations would yield stricter quantitative formulation of these differences for the specific systems.
The phonon frequencies obtained by ab initio calculations is to be used for assessing the thermodynamic properties of layers and nanotubes (by summing the relevant contributions over set of BZ points) at different temperatures. These computations allow the direct comparison of thermodynamic properties (Helmholtz energy, entropy and heat capacity) of flat layers and nanotubes obtained by their folding. Knowledge of free energies will enable the correct estimate of the nanotube relative stability and study its temperature dependence. The calculation of thermal expansion coefficients, as well as modules of the elasticity of layers and nanotubes is also planned.
Relevant publications
- A. V. Bandura, V. V. Porsev, R. A. Evarestov, Application of Zone-Folding Approach to the First-Principles Estimation of Thermodynamic Properties of Carbon and ZrS2-Based Nanotubes. J. Comput. Chem. 2016, 37, 641–652. DOI: http://dx.doi.org/10.1002/jcc.24243.
- A. V. Bandura, D. D. Kuruch, R. A. Evarestov, First-principles Calculations of InS-based Nanotubes. Isr. J. Chem. 2017, 57, 490– 500. DOI: http://dx.doi.org/10.1002/ijch.201600054
- R. A. Evarestov, A. V. Bandura, V. V. Porsev, A. V. Kovalenko, Phonon Spectra, Electronic, and Thermodynamic Properties of WS2 Nanotubes. J. Comput. Chem. 2017, in print. DOI: http://dx.doi.org/10.1002/jcc.24916
Figure Captions
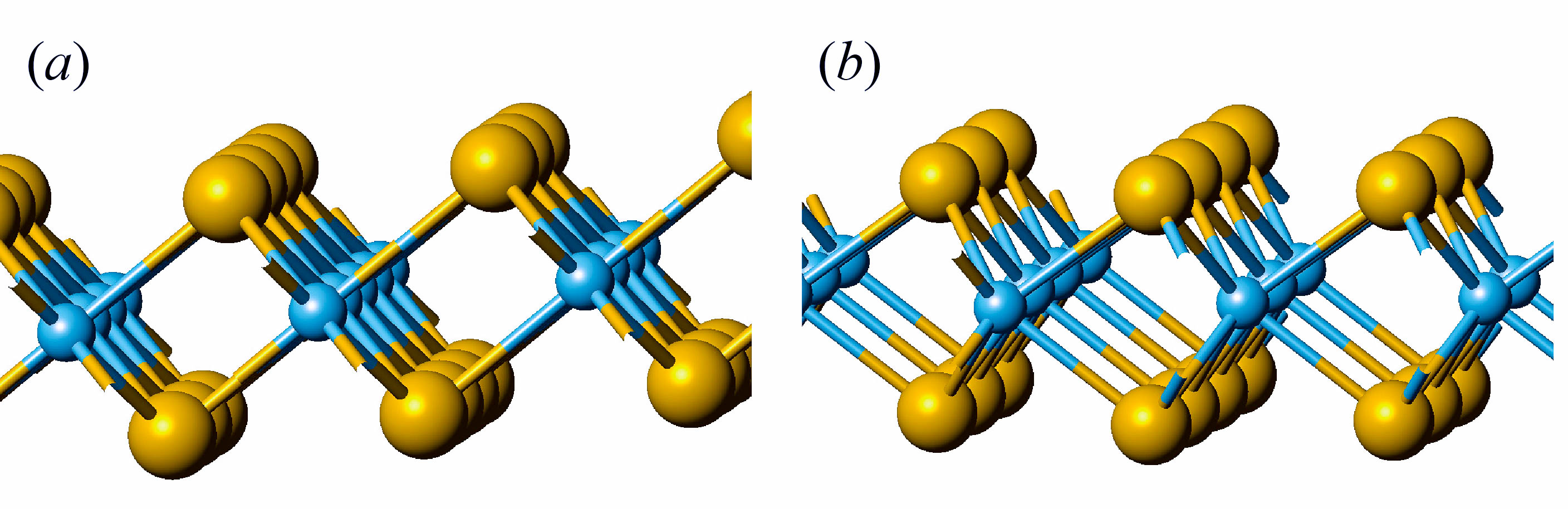
Fig. 1. Monolayer structure of ZrS2 with hexagonal Zr coordination (a), and MoS2 with trigonal-prismatic coordination of Mo (b). Sulfur atoms – large spheres, metal atoms – small spheres.
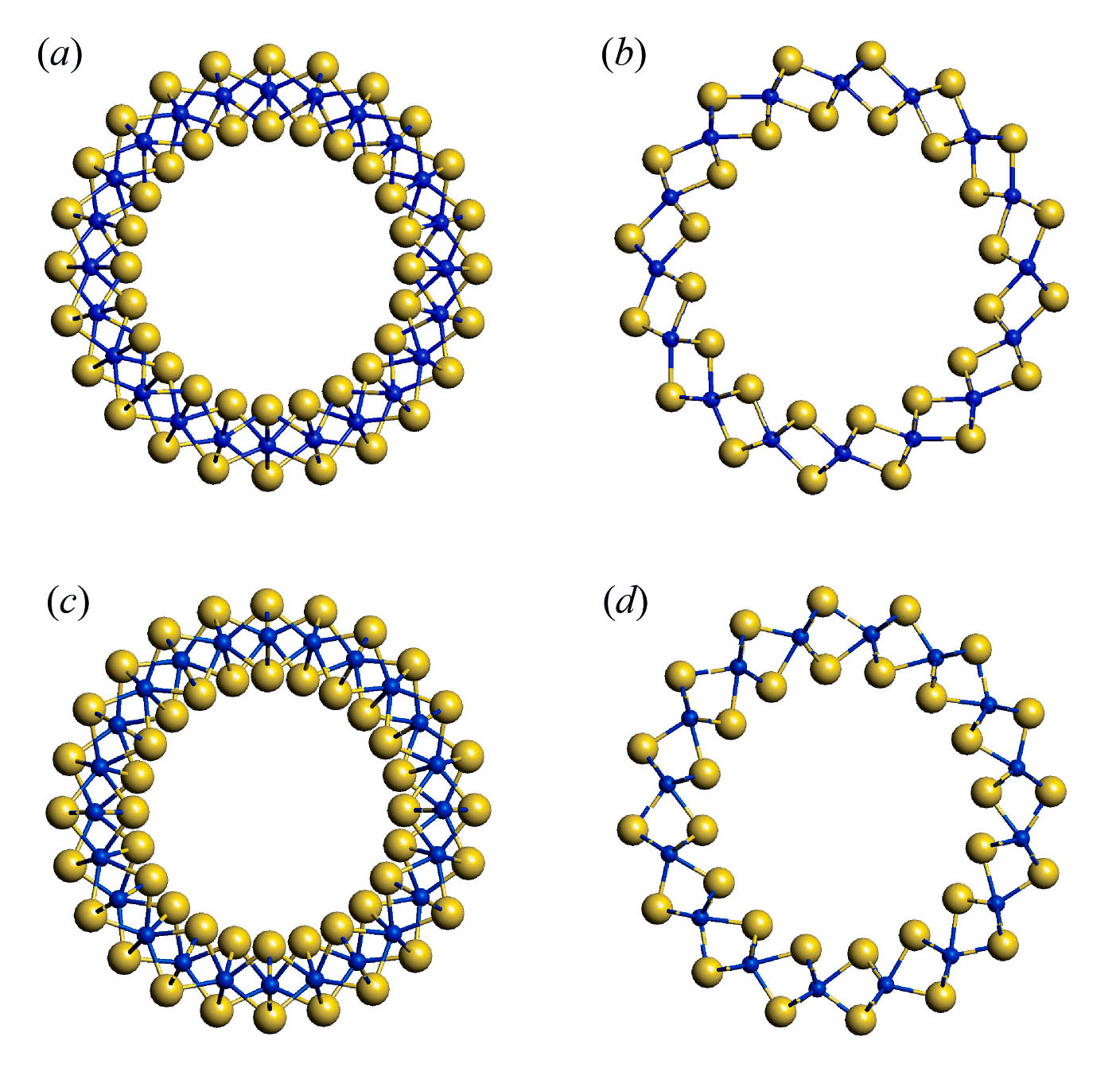
Fig. 2. Cross-section view of nanotubes obtained by rolling up of monolayers of ZrS2 (a, b) and MoS2 (c, d) with chirality (12, 0) (a, c) and (8, 8) (b, d). Sulfur atoms – large spheres, metal atoms – small spheres.